Thermodynamics Cheatsheet
Cheatsheet Content
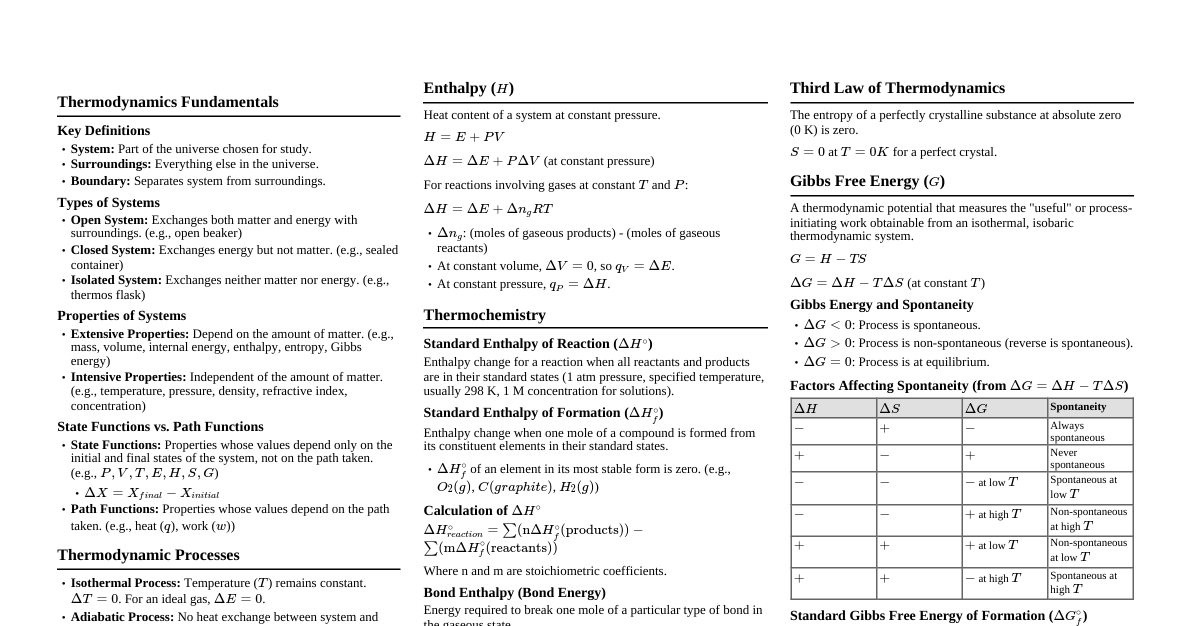
1. Fundamental Concepts Thermodynamic Terms System: The specific part of the universe under observation. Surroundings: Everything in the universe except the system. Boundary: The real or imaginary wall separating the system from the surroundings. Types of Systems: Open System: Can exchange both energy and matter with the surroundings. Closed System: Can exchange energy but not matter. Isolated System: Can exchange neither energy nor matter. State Functions and Path Functions State Functions (or State Variables): Properties whose values depend only on the initial and final states of the system, not on the path taken to reach that state. Examples: Internal Energy ($\Delta U$), Enthalpy ($\Delta H$), Entropy ($\Delta S$), Gibbs Free Energy ($\Delta G$), Pressure, Volume, Temperature. Path Functions: Properties whose values depend on the path followed by the system. Examples: Work ($W$), Heat ($q$). Thermodynamic Processes Isothermal Process: Temperature remains constant ($dT = 0$, $\Delta U = 0$ for an ideal gas). Isobaric Process: Pressure remains constant ($dP = 0$). Isochoric Process: Volume remains constant ($dV = 0$). Adiabatic Process: No heat is exchanged with the surroundings ($q = 0$). Reversible Process: A process conducted in such a way that it can be reversed by an infinitesimal change in conditions. It proceeds through a series of equilibrium states. Irreversible Process: A process that cannot be reversed. All natural processes are irreversible. The First Law of Thermodynamics This law is based on the principle of conservation of energy, stating that energy can neither be created nor destroyed, only converted from one form to another. Mathematically, it is expressed as: $\Delta U = q + W$ Where: $\Delta U$ = Change in internal energy of the system. $q$ = Heat supplied to the system. $W$ = Work done on the system. Sign Conventions: $q$ is positive ($+$) when heat is supplied to the system. $W$ is positive ($+$) when work is done on the system (compression). $q$ is negative ($-$) when heat is lost by the system. $W$ is negative ($-$) when work is done by the system (expansion). 2. Work, Heat, and Enthalpy Pressure-Volume Work Work in irreversible expansion (against constant external pressure $p_{ext}$): $W = -p_{ext}\Delta V = -p_{ext}(V_2 - V_1)$ Work in isothermal reversible expansion: $W_{rev} = -2.303nRT \log\left(\frac{V_2}{V_1}\right) = -2.303nRT \log\left(\frac{P_1}{P_2}\right)$ Work in adiabatic reversible expansion: $W_{rev} = \frac{nR}{\gamma - 1}(T_2 - T_1)$ where $\gamma = C_p/C_v$ is the Poisson's ratio. Heat Capacity Heat Capacity (C) is the heat required to raise a system's temperature by $1^\circ C$. Specific Heat Capacity (c): Heat capacity per gram of substance. $q = mc\Delta T$. Molar Heat Capacity (C$_m$): Heat capacity per mole of substance. For an ideal gas, the molar heat capacities at constant pressure ($C_p$) and constant volume ($C_v$) are related by: $C_p - C_v = R$ Enthalpy (H) Enthalpy is the total heat content of a system. It is the sum of the internal energy and the pressure-volume energy. It is a state function and an extensive property. $H = U + pV$ The change in enthalpy ($\Delta H$) is equal to the heat absorbed or evolved by the system at constant pressure. $\Delta H = q_p$ Relationship Between Enthalpy and Internal Energy $\Delta H = \Delta U + p\Delta V$ For reactions involving gases, this can be expressed as: $\Delta H = \Delta U + \Delta n_g RT$ where $\Delta n_g$ is the change in the number of moles of gaseous products and reactants. Types of Reaction Enthalpies ($\Delta H$) Enthalpy of Formation ($\Delta_f H^\circ$): Enthalpy change when one mole of a compound is formed from its constituent elements in their standard states. By convention, $\Delta_f H^\circ$ of elements in their standard state is zero. Enthalpy of Combustion ($\Delta_c H^\circ$): Enthalpy change when one mole of a substance undergoes complete combustion. This process is always exothermic ($\Delta_c H^\circ$ is negative). Enthalpy of Neutralisation ($\Delta_{neut} H^\circ$): Enthalpy change when 1 g-equivalent of an acid is completely neutralized by 1 g-equivalent of a base in dilute solution. For a strong acid and strong base, this value is constant at approximately $-57.1 \text{ kJ/mol}$. Enthalpy of Atomisation ($\Delta_{atom} H^\circ$): Enthalpy change when one mole of a substance breaks into its constituent atoms in the gaseous phase. Bond Enthalpy: The energy required to break one mole of a specific type of bond in a gaseous molecule. Lattice Enthalpy: The enthalpy change required to completely separate one mole of a solid ionic compound into its gaseous constituent ions. 3. Laws of Thermochemistry and Spontaneity Hess's Law of Constant Heat Summation The total enthalpy change for a chemical reaction is the same, regardless of whether the reaction occurs in one step or in a series of steps. This allows for the calculation of enthalpy changes for reactions that cannot be measured directly. $\Delta H_{reaction} = \Sigma \Delta H_{steps}$ Important Thermochemical Laws Kirchhoff's Equation: Relates the change in reaction enthalpy with temperature. $\Delta H_{T_2} - \Delta H_{T_1} = \Delta C_p(T_2 - T_1)$ Trouton's Rule: For many liquids, the entropy of vaporisation has a similar value. $\frac{\Delta H_{vap}}{T_b} \approx 88 \text{ J K}^{-1}\text{mol}^{-1}$ Clausius-Clapeyron Equation: Relates the vapor pressure of a liquid to its temperature. $\log\frac{P_2}{P_1} = \frac{\Delta H_{vap}}{2.303R}\left(\frac{T_2 - T_1}{T_1 T_2}\right)$ Spontaneous Process A process that can proceed on its own without any external assistance under a given set of conditions. All natural processes are spontaneous and irreversible. Spontaneity is not necessarily related to the speed of the process. Entropy (S) Entropy is a measure of the degree of randomness or disorder in a system. It is a state function and an extensive property. The entropy of the universe always increases during a spontaneous process. $\Delta S = \frac{q_{rev}}{T}$ The criterion for a spontaneous process is an increase in the total entropy of the universe: $\Delta S_{total} = \Delta S_{system} + \Delta S_{surroundings} > 0$ 4. Gibbs Free Energy and Thermodynamic Laws The Second and Third Laws of Thermodynamics Second Law of Thermodynamics: The entropy of the universe is always increasing in the course of every spontaneous (natural) change. It also implies that heat cannot spontaneously flow from a colder body to a hotter body. Third Law of Thermodynamics: The entropy of a perfectly crystalline substance at absolute zero (0 K) is taken to be zero. Gibbs Free Energy (G) Gibbs free energy is the portion of a system's total energy that is available to do useful work. It is a state function and an extensive property. $G = H - TS$ Gibbs Free Energy and Spontaneity The change in Gibbs free energy ($\Delta G$) for a process at constant temperature is given by the Gibbs-Helmholtz equation: $\Delta G = \Delta H - T\Delta S$ The sign of $\Delta G$ is the ultimate criterion for spontaneity at constant temperature and pressure: If $\Delta G spontaneous. If $\Delta G > 0$: The process is non-spontaneous. If $\Delta G = 0$: The system is in equilibrium. Effect of Temperature on Spontaneity $\Delta H$ $\Delta S$ $\Delta G = \Delta H - T\Delta S$ Outcome $-$ $+$ Always negative Spontaneous at all T $+$ $-$ Always positive Non-spontaneous at all T $-$ $-$ Becomes negative at high T Spontaneous at high T $+$ $+$ Becomes negative at low T Spontaneous at low T Gibbs Free Energy and Equilibrium The standard free energy change ($\Delta G^\circ$) is related to the equilibrium constant ($K$) of a reaction: $\Delta G^\circ = -RT \ln K = -2.303RT \log_{10} K$ It is also related to the standard cell potential ($E_{cell}$) of an electrochemical cell: $\Delta G^\circ = -nFE_{cell}^\circ$ Where $n$ is the number of moles of electrons transferred and $F$ is the Faraday constant ($96500 \text{ C/mol}$).