Chemistry Essentials
Cheatsheet Content
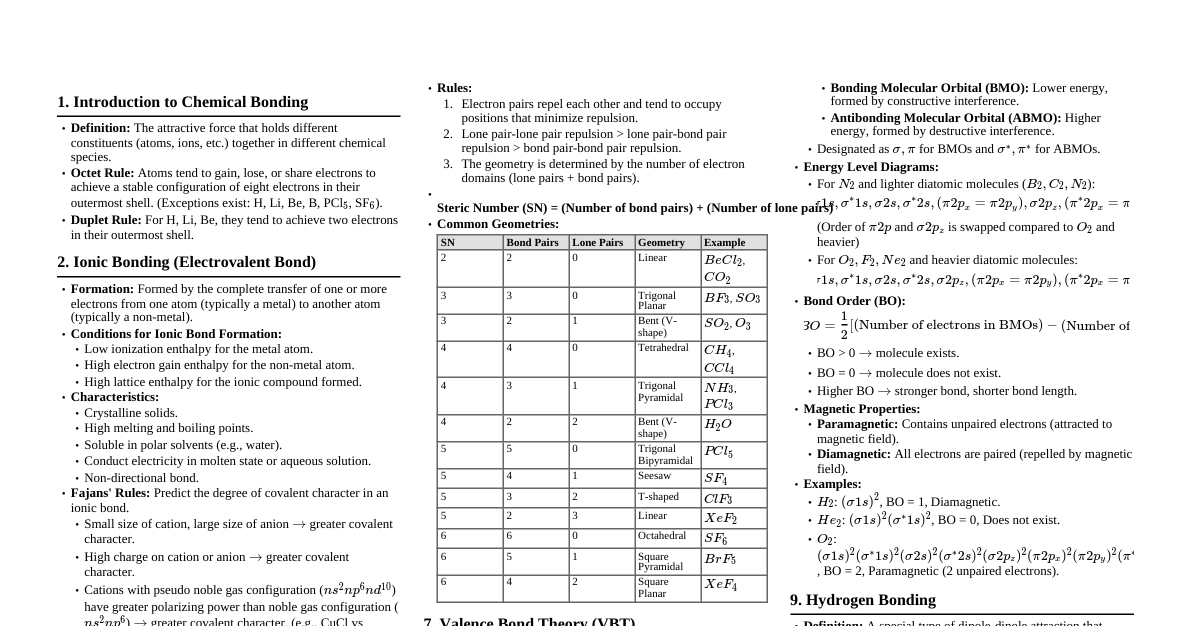
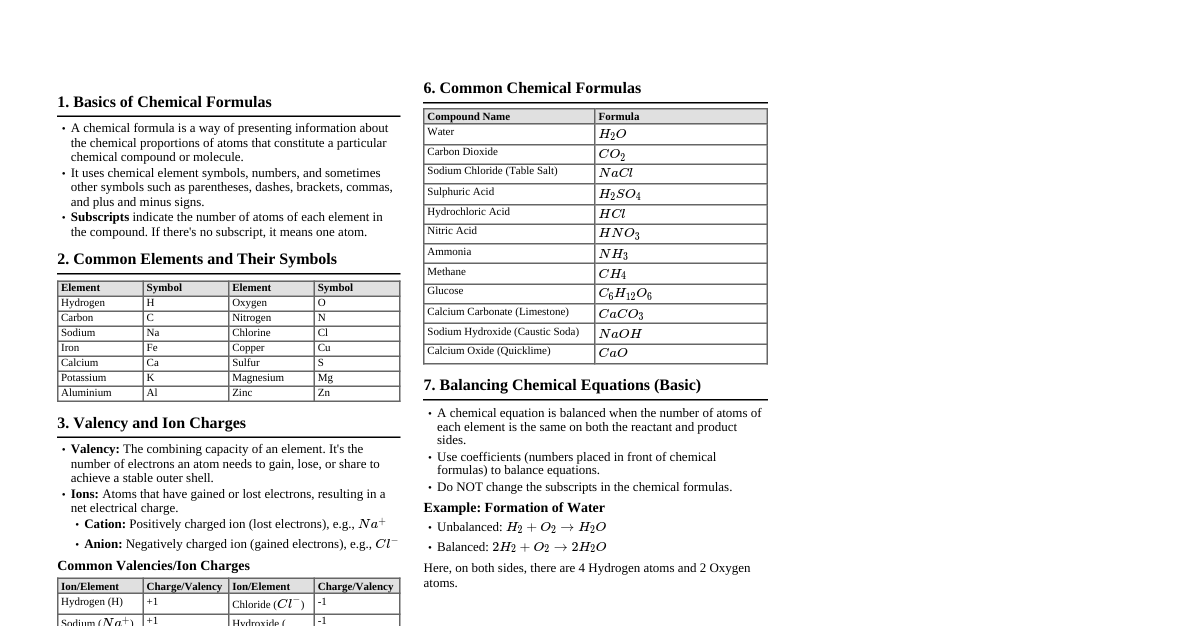
Chemical Bonding & Molecular Structure 1. Kössel-Lewis Approach Octet Rule: Atoms tend to achieve a stable outer shell of 8 electrons (octet) by forming chemical bonds. Exceptions: H (duplet), elements in Period 3 and beyond (expanded octet). Lewis Symbols: Dots around an element symbol represent valence electrons. Example: $\cdot \text{Li}$, $\cdot \text{Be} \cdot$, $: \text{O} :$ Ionic Bond: Transfer of electrons, forming ions. Electrostatic attraction between ions. Example: $\text{Na} \to \text{Na}^+ + \text{e}^-$, $\text{Cl} + \text{e}^- \to \text{Cl}^-$ Electrovalence: Number of unit charges on the ion. Covalent Bond: Sharing of electrons between atoms. Single Bond: 1 shared pair (e.g., $\text{H}-\text{H}$) Double Bond: 2 shared pairs (e.g., $\text{O}=\text{O}$) Triple Bond: 3 shared pairs (e.g., $\text{N} \equiv \text{N}$) Lewis Dot Structures: Represent shared and lone pairs. Formal Charge (FC): $\text{FC} = (\text{Valence electrons}) - (\text{Non-bonding electrons}) - \frac{1}{2}(\text{Bonding electrons})$ 2. VSEPR Theory (Valence Shell Electron Pair Repulsion) Predicts molecular geometry based on minimizing electron pair repulsion. Repulsion Order: $\text{Lone Pair-Lone Pair (LP-LP)} > \text{Lone Pair-Bond Pair (LP-BP)} > \text{Bond Pair-Bond Pair (BP-BP)}$ Summary of Geometries (No Lone Pairs on Central Atom): Electron Pairs Arrangement Geometry Examples 2 Linear Linear $\text{BeCl}_2$ 3 Trigonal Planar Trigonal Planar $\text{BF}_3$ 4 Tetrahedral Tetrahedral $\text{CH}_4$ 5 Trigonal Bipyramidal Trigonal Bipyramidal $\text{PCl}_5$ 6 Octahedral Octahedral $\text{SF}_6$ Summary of Geometries (With Lone Pairs on Central Atom): Bonding Pairs Lone Pairs Arrangement Shape Examples 2 1 Trigonal Planar Bent $\text{SO}_2$ 3 1 Tetrahedral Trigonal Pyramidal $\text{NH}_3$ 2 2 Tetrahedral Bent $\text{H}_2\text{O}$ 4 1 Trigonal Bipyramidal See-saw $\text{SF}_4$ 3 2 Trigonal Bipyramidal T-shape $\text{ClF}_3$ 5 1 Octahedral Square Pyramidal $\text{BrF}_5$ 4 2 Octahedral Square Planar $\text{XeF}_4$ 3. Valence Bond Theory (VBT) Covalent bonds form by overlapping atomic orbitals. Greater overlap = stronger bond. Types of Overlap: Sigma ($\sigma$) Bond: Head-on (axial) overlap. Stronger. s-s overlap: $\text{H}_2$ s-p overlap: $\text{HCl}$ p-p overlap: $\text{Cl}_2$ (along internuclear axis) Pi ($\pi$) Bond: Sideways overlap of p-orbitals. Weaker than $\sigma$. Formed in addition to a $\sigma$ bond in multiple bonds. Hybridization: Mixing of atomic orbitals to form new equivalent hybrid orbitals. Number of hybrid orbitals = number of atomic orbitals mixed. Hybrid orbitals are equivalent in energy and shape. Conditions: Valence shell orbitals, similar energy, maximum overlap. Types: $\text{sp}$: 1 s + 1 p $\to$ 2 sp orbitals. Linear geometry. Example: $\text{BeCl}_2$. $\text{sp}^2$: 1 s + 2 p $\to$ 3 sp$^2$ orbitals. Trigonal planar. Example: $\text{BF}_3$. $\text{sp}^3$: 1 s + 3 p $\to$ 4 sp$^3$ orbitals. Tetrahedral. Example: $\text{CH}_4$. Involving d-orbitals: $\text{sp}^3\text{d}$ (trigonal bipyramidal, $\text{PCl}_5$), $\text{sp}^3\text{d}^2$ (octahedral, $\text{SF}_6$) 4. Molecular Orbital Theory (MOT) Atomic orbitals combine to form molecular orbitals (MOs). Number of MOs = number of atomic orbitals combined. Bonding MOs: Lower energy, stable (electron density between nuclei). Antibonding MOs: Higher energy, unstable (electron density away from nuclei). Bond Order (BO): $\text{BO} = \frac{1}{2} (\text{N}_b - \text{N}_a)$. $\text{N}_b$ = electrons in bonding MOs, $\text{N}_a$ = electrons in antibonding MOs. $\text{BO} > 0 \implies$ stable molecule. $\text{BO} = 0 \implies$ unstable molecule. Magnetic Properties: Paramagnetic: Unpaired electrons in MOs (attracted to magnetic field). Diamagnetic: All electrons paired in MOs (repelled by magnetic field). Energy Order of MOs: For $\text{O}_2, \text{F}_2$: $\sigma1\text{s} For $\text{Li}_2 \text{ to } \text{N}_2$: $\sigma1\text{s} 5. Hydrogen Bonding Attractive force between $\text{H}$ atom (bonded to highly electronegative atom like $\text{F}, \text{O}, \text{N}$) and another electronegative atom. Weaker than covalent bonds. Represented by a dotted line (--). Intermolecular H-bond: Between different molecules (e.g., $\text{H}_2\text{O}$). Intramolecular H-bond: Within the same molecule (e.g., o-nitrophenol). Thermodynamics 1. Basic Concepts System: Part of the universe under study. Surroundings: Rest of the universe. Types of Systems: Open: Exchange of matter and energy (e.g., beaker of water). Closed: Exchange of energy, no matter (e.g., sealed container). Isolated: No exchange of matter or energy (e.g., thermos flask). State Function: Property whose value depends only on the state of the system, not on how it was reached (e.g., $U, H, S, G, T, P, V$). Path Function: Property whose value depends on the path taken (e.g., $q, w$). Internal Energy ($U$): Total energy of the system. A state function. Change in internal energy ($\Delta U$) can occur via heat ($q$) or work ($w$). Work ($w$): Energy transferred due to force acting over a distance. Pressure-Volume Work: $w = -p_{\text{ex}}\Delta V$. Work done on system (compression): $w > 0$. Work done by system (expansion): $w Reversible Work (Isothermal): $w_{\text{rev}} = -nRT \ln \frac{V_f}{V_i} = -2.303 nRT \log \frac{V_f}{V_i}$. Free Expansion: $p_{\text{ex}} = 0 \implies w = 0$. Heat ($q$): Energy transferred due to temperature difference. Heat absorbed by system: $q > 0$. Heat released by system: $q 2. First Law of Thermodynamics Statement: Energy can neither be created nor destroyed. The energy of an isolated system is constant. Mathematical Form: $\Delta U = q + w$. Adiabatic Process: $q = 0 \implies \Delta U = w_{\text{ad}}$. Isothermal Process: $\Delta U = 0$ (for ideal gas) $\implies q = -w$. Constant Volume Process: $\Delta V = 0 \implies w = 0 \implies \Delta U = q_V$. 3. Enthalpy ($H$) Definition: $H = U + pV$. A state function. Change in Enthalpy: $\Delta H = \Delta U + p\Delta V$ (at constant pressure). Constant Pressure Process: $\Delta H = q_p$. Relation between $\Delta H$ and $\Delta U$ (for gases): $\Delta H = \Delta U + \Delta n_g RT$. $\Delta n_g = (\text{moles of gaseous products}) - (\text{moles of gaseous reactants})$. Heat Capacity ($C$): Amount of heat required to raise temperature by $1^\circ \text{C}$ (or $1 \text{ K}$). $q = C\Delta T$. Molar Heat Capacity ($C_m$): Per mole of substance. Specific Heat Capacity ($c$): Per unit mass of substance. $q = mc\Delta T$. Relation between $C_p$ and $C_V$ (for ideal gas): $C_p - C_V = R$. Extensive Properties: Depend on quantity of matter (e.g., $U, H, V$, mass, heat capacity). Intensive Properties: Independent of quantity of matter (e.g., $T, P$, density, molar heat capacity). 4. Reaction Enthalpies Reaction Enthalpy ($\Delta_r H$): Enthalpy change for a reaction. $\Delta_r H = \sum H_{\text{products}} - \sum H_{\text{reactants}}$. Standard State: Pure form at 1 bar pressure and specified temperature (usually 298 K). Denoted by $\Delta H^\circ$. Standard Enthalpy of Formation ($\Delta_f H^\circ$): Enthalpy change when 1 mole of a compound is formed from its elements in their most stable states at standard conditions. $\Delta_f H^\circ (\text{element in standard state}) = 0$. $\Delta_r H^\circ = \sum a_i \Delta_f H^\circ (\text{products}) - \sum b_i \Delta_f H^\circ (\text{reactants})$. Hess's Law of Constant Heat Summation: The total enthalpy change for a reaction is the same regardless of the path taken (i.e., whether it occurs in one step or several steps). Standard Enthalpy of Combustion ($\Delta_c H^\circ$): Enthalpy change when 1 mole of a substance undergoes complete combustion at standard conditions. Enthalpy of Atomization ($\Delta_a H^\circ$): Enthalpy change to break all bonds in 1 mole of substance to form gaseous atoms. Bond Enthalpy ($\Delta_{\text{bond}} H^\circ$): Energy required to break a specific bond in the gas phase. Mean bond enthalpy is used for polyatomic molecules. $\Delta_r H^\circ \approx \sum (\text{bond enthalpies of reactants}) - \sum (\text{bond enthalpies of products})$. Lattice Enthalpy ($\Delta_{\text{lattice}} H^\circ$): Enthalpy change when 1 mole of an ionic compound dissociates into its gaseous ions. Calculated using Born-Haber cycle. Enthalpy of Solution ($\Delta_{\text{sol}} H^\circ$): Enthalpy change when 1 mole of substance dissolves in a specified amount of solvent. $\Delta_{\text{sol}} H^\circ = \Delta_{\text{lattice}} H^\circ + \Delta_{\text{hyd}} H^\circ$. Enthalpy of Dilution: Heat change when additional solvent is added to a solution. 5. Spontaneity Spontaneous Process: Occurs without external assistance. Irreversible. Does not imply fast rate. Entropy ($S$): Measure of disorder or randomness. A state function. $\Delta S = \frac{q_{\text{rev}}}{T}$. For a spontaneous process in an isolated system: $\Delta S_{\text{total}} = \Delta S_{\text{sys}} + \Delta S_{\text{surr}} > 0$. At equilibrium: $\Delta S_{\text{total}} = 0$. Third Law of Thermodynamics: Entropy of a pure crystalline substance approaches zero as $T \to 0 \text{ K}$. Gibbs Energy ($G$): A thermodynamic potential that measures useful or process-initiating work obtainable from an isothermal, isobaric thermodynamic system. A state function. Definition: $G = H - TS$. Change in Gibbs Energy: $\Delta G = \Delta H - T\Delta S$ (at constant $T, P$). Criteria for Spontaneity (at constant $T, P$): $\Delta G $\Delta G > 0 \implies$ Non-spontaneous. $\Delta G = 0 \implies$ At equilibrium. Effect of $T$ on Spontaneity: $\Delta H$ $\Delta S$ $\Delta G = \Delta H - T\Delta S$ Spontaneity $-$ $+$ $-$ Spontaneous at all $T$ $-$ $-$ $-$ (low $T$) Spontaneous at low $T$ $-$ $-$ $+$ (high $T$) Non-spontaneous at high $T$ $+$ $+$ $+$ (low $T$) Non-spontaneous at low $T$ $+$ $+$ $-$ (high $T$) Spontaneous at high $T$ $+$ $-$ $+$ Non-spontaneous at all $T$ 6. Gibbs Energy Change and Equilibrium Relation to Equilibrium Constant ($K$): $\Delta G^\circ = -RT \ln K = -2.303 RT \log K$. If $\Delta G^\circ 1$, products favored. If $\Delta G^\circ > 0$, $K If $\Delta G^\circ = 0$, $K = 1$, at equilibrium.