Chemistry Thermodynamics (NEET)
Cheatsheet Content
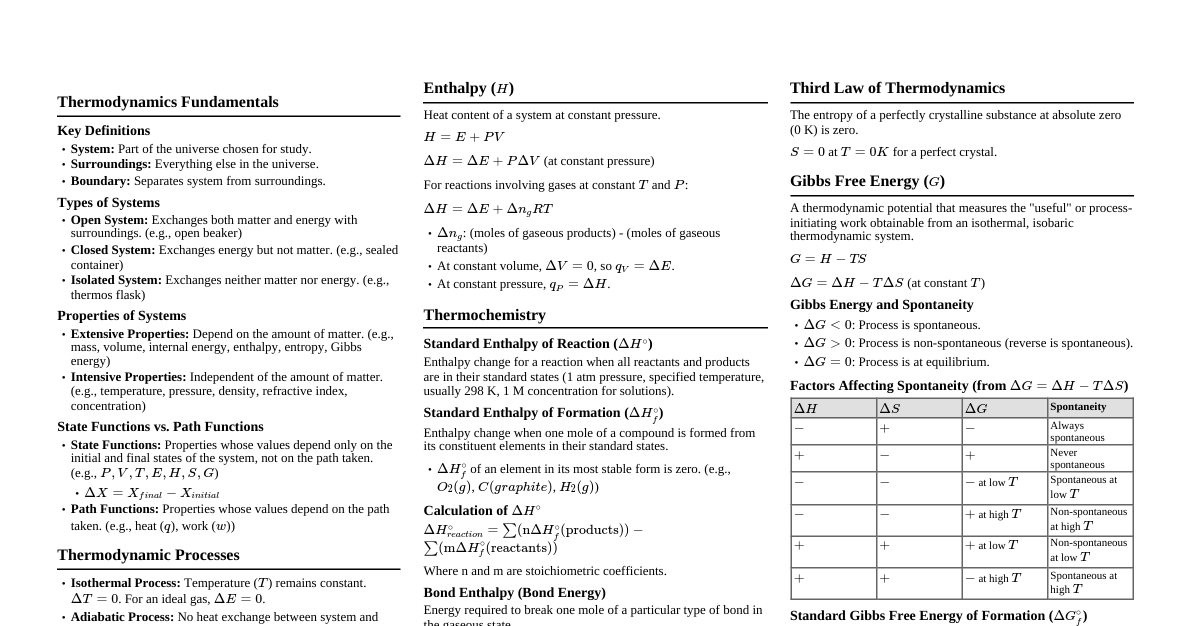
1. Introduction to Thermodynamics System: Part of the universe chosen for study. Surroundings: Everything else in the universe. Boundary: Separates system from surroundings. Types of Systems: Open: Exchanges both matter and energy. Closed: Exchanges energy but not matter. Isolated: Exchanges neither matter nor energy. State Functions: Properties that depend only on the initial and final states, not on the path (e.g., $P, V, T, U, H, S, G$). Path Functions: Properties that depend on the path taken (e.g., $q, w$). Extensive Properties: Depend on the amount of matter (e.g., $V, U, H, S, G$). Intensive Properties: Independent of the amount of matter (e.g., $P, T, \text{density}$). 2. First Law of Thermodynamics Statement: Energy can neither be created nor destroyed, only transformed. Mathematical Form: $\Delta U = q + w$ $\Delta U$: Change in internal energy. $q$: Heat absorbed by the system (positive if absorbed, negative if released). $w$: Work done on the system (positive if on system, negative if by system). Work Done ($w$): Pressure-Volume Work: $w = -P_{ext}\Delta V$ (for irreversible process) Reversible Work: $w = -nRT \ln \frac{V_2}{V_1} = -nRT \ln \frac{P_1}{P_2}$ (for isothermal expansion/compression of ideal gas) Heat Capacity ($C$): $q = C\Delta T$ $C_v$ (at constant volume): $\Delta U = q_v = nC_v\Delta T$ $C_p$ (at constant pressure): $\Delta H = q_p = nC_p\Delta T$ Relation: $C_p - C_v = R$ (for ideal gas) $\gamma = \frac{C_p}{C_v}$ 3. Enthalpy ($H$) Definition: $H = U + PV$ Change in Enthalpy: $\Delta H = \Delta U + P\Delta V$ (at constant pressure) Relation between $\Delta H$ and $\Delta U$: $\Delta H = \Delta U + \Delta n_g RT$ $\Delta n_g$: Change in moles of gaseous products minus gaseous reactants. Types of Enthalpies: Standard Enthalpy of Formation ($\Delta H_f^\circ$): Enthalpy change when 1 mole of a compound is formed from its elements in their standard states. $\Delta H_f^\circ (\text{element}) = 0$. Standard Enthalpy of Reaction ($\Delta H_r^\circ$): $\Delta H_r^\circ = \sum \Delta H_f^\circ (\text{products}) - \sum \Delta H_f^\circ (\text{reactants})$ Bond Enthalpy: Energy required to break one mole of a specific bond. $\Delta H_r^\circ = \sum (\text{Bond Enthalpies of reactants}) - \sum (\text{Bond Enthalpies of products})$ Enthalpy of Combustion ($\Delta H_c^\circ$): Heat released when 1 mole of substance is completely burnt in oxygen. Enthalpy of Neutralization: Heat released when 1 mole of $H^+$ reacts with 1 mole of $OH^-$ to form water. Enthalpy of Solution: Heat change when 1 mole of a substance dissolves in a specified amount of solvent. Enthalpy of Fusion/Vaporization: Heat change during phase transitions. Hess's Law: The total enthalpy change for a reaction is the same, regardless of the number of steps or path taken. 4. Second Law of Thermodynamics Statement: The entropy of an isolated system always increases in the course of a spontaneous change. Entropy ($S$): Measure of randomness or disorder. Change in Entropy: $\Delta S = \frac{q_{rev}}{T}$ $\Delta S_{sys} = \sum \frac{q}{T}$ $\Delta S_{total} = \Delta S_{sys} + \Delta S_{surr}$ For spontaneous process: $\Delta S_{total} > 0$ For equilibrium: $\Delta S_{total} = 0$ Standard Molar Entropy ($S^\circ$): Entropy of 1 mole of a substance at standard conditions. $S^\circ$ for elements is not zero. Entropy of Reaction: $\Delta S_r^\circ = \sum S^\circ (\text{products}) - \sum S^\circ (\text{reactants})$ Factors increasing entropy: Increase in temperature, volume, number of moles of gas, phase change from solid to liquid to gas. 5. Third Law of Thermodynamics Statement: The entropy of a perfectly crystalline substance at absolute zero (0 K) is taken as zero. 6. Gibbs Free Energy ($G$) Definition: $G = H - TS$ Change in Gibbs Free Energy: $\Delta G = \Delta H - T\Delta S$ (at constant $T, P$) Predicting Spontaneity: $\Delta G $\Delta G > 0$: Non-spontaneous process (reverse is spontaneous) $\Delta G = 0$: System is at equilibrium Effect of $\Delta H$ and $\Delta S$ on Spontaneity: $\Delta H$ $\Delta S$ $\Delta G = \Delta H - T\Delta S$ Spontaneity $-$ $+$ Always $-$ Spontaneous at all $T$ $+$ $-$ Always $+$ Non-spontaneous at all $T$ $-$ $-$ $-$ at low $T$, $+$ at high $T$ Spontaneous at low $T$ $+$ $+$ $+$ at low $T$, $-$ at high $T$ Spontaneous at high $T$ Relation to Equilibrium Constant ($K$): $\Delta G^\circ = -RT \ln K$ $\Delta G = \Delta G^\circ + RT \ln Q$ (where $Q$ is reaction quotient) At equilibrium, $\Delta G = 0$, so $\Delta G^\circ = -RT \ln K$ Standard Gibbs Free Energy of Formation ($\Delta G_f^\circ$): $\Delta G_f^\circ (\text{element}) = 0$. Gibbs Free Energy of Reaction: $\Delta G_r^\circ = \sum \Delta G_f^\circ (\text{products}) - \sum \Delta G_f^\circ (\text{reactants})$ 7. Important Processes Isothermal: $\Delta T = 0$. For ideal gas, $\Delta U = 0$, $\Delta H = 0$. $q = -w$. Adiabatic: $q = 0$. $\Delta U = w$. Isobaric: $\Delta P = 0$. $q = \Delta H$. Isochoric: $\Delta V = 0$. $w = 0$. $q = \Delta U$. Cyclic Process: $\Delta U = 0$, $\Delta H = 0$, $\Delta S = 0$, $\Delta G = 0$. 8. Key Formulas Summary $\Delta U = q + w$ $w = -P_{ext}\Delta V$ $\Delta H = \Delta U + \Delta n_g RT$ $\Delta S = \frac{q_{rev}}{T}$ $\Delta G = \Delta H - T\Delta S$ $\Delta G^\circ = -RT \ln K$ $C_p - C_v = R$ $\Delta H_r^\circ = \sum \Delta H_f^\circ (\text{products}) - \sum \Delta H_f^\circ (\text{reactants})$ $\Delta S_r^\circ = \sum S^\circ (\text{products}) - \sum S^\circ (\text{reactants})$ $\Delta G_r^\circ = \sum \Delta G_f^\circ (\text{products}) - \sum \Delta G_f^\circ (\text{reactants})$